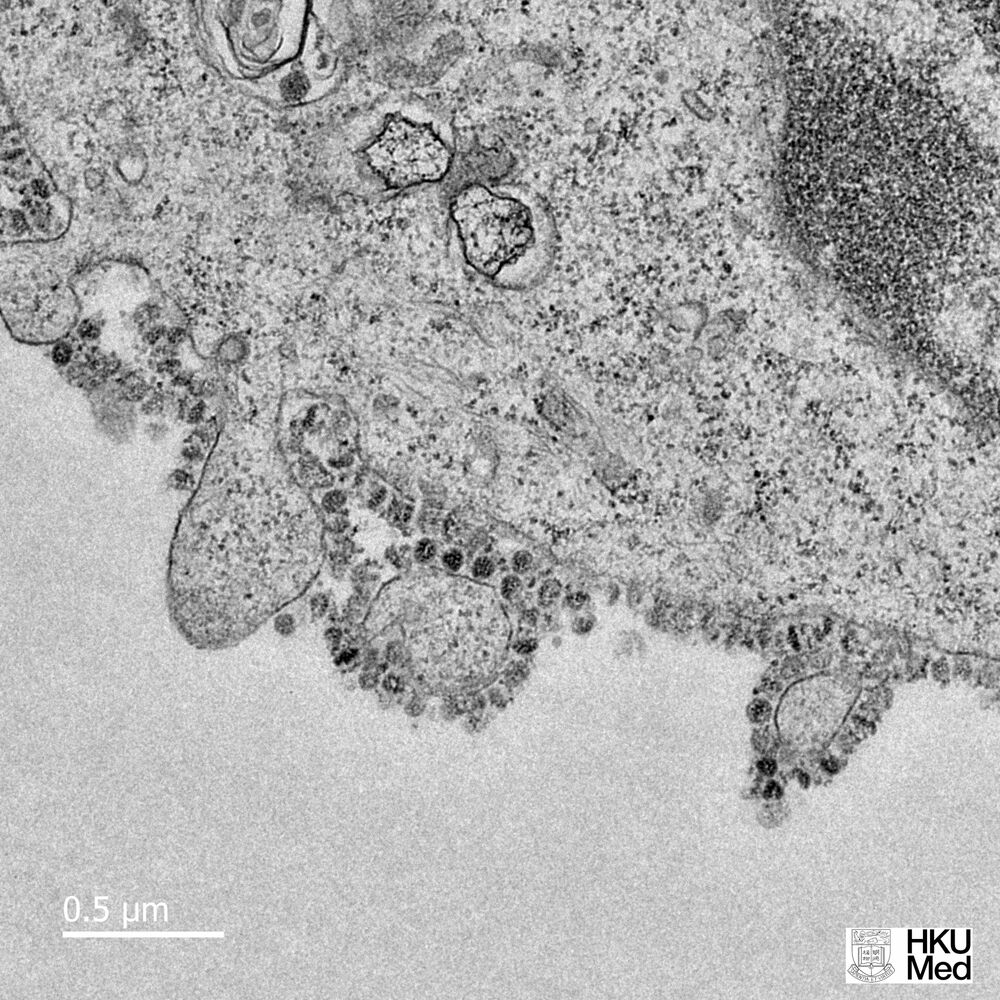
In a month where all academic and professional examinations have been cancelled or postponed, on the 16th of March, the clarion call from the leader of the World Health Organisation, Tedros Adhanom Ghebreyesus was "test, test, test!". In the last few days, a major focus of news broadcasts has been the controversy surrounding the number of tests carried out per day, the countries who are prioritising testing, the reliability of the forecasts that influence government advice and regulation of its citizens, as well as the management of NHS and social care resources. Pretty important stuff, I think you would all agree. Since Science lies at the heart of this series of Blog posts, I thought I would also prioritise testing as a topic over the basic of antiviral therapies (which will follow soon).
In a month where all academic and professional examinations have been cancelled or postponed, on the 16th of March, the clarion call from the leader of the World Health Organisation, Tedros Adhanom Ghebreyesus was "test, test, test!". In the last few days, a major focus of news broadcasts has been the controversy surrounding the number of tests carried out per day, the countries who are prioritising testing, the reliability of the forecasts that influence government advice and regulation of its citizens, as well as the management of NHS and social care resources. Pretty important stuff, I think you would all agree. Since Science lies at the heart of this series of Blog posts, I thought I would also prioritise testing as a topic over the basic of antiviral therapies (which will follow soon).At this point, you may wish to jump to the section below highlighted in red, if you are familiar with the basics of analytical chemistry.
What do we mean by a "test"? Usually we mean one of two things:
1. An instrument (used in the academic sense, it might be a series of questions or exercises, for example) for measuring skill, knowledge, intelligence, capacity or aptitude of an individual or a group.
or in this case:
2. A procedure or a reaction combining a set of reagents, used to identify a particular substance (or class of substances) in a sample.
Working with the somewhat unsatisfactory vagueness of the second definition, let me begin by discussing the principles of testing, which have their roots in Analytical Chemistry, and then get to the specifics of the tests in the media, where terms and acronyms like "antibody test", "antigen test", "PCR" test and "virus test", are being bandied around.
Sensitive (sufficient to detect amounts of a substance that give rise to a problem: in the case of arsenic, it is necessary to detect levels of arsenic that are at least the amounts that are damaging to health). [I shall refer later to the concept of the "signal to noise ratio"].
Specific (arsenic [As] appears in Group 15, period 4 of the periodic table, below Phosphorus and above Antimony: any test must make an unequivocal distinction between these closely related elements)
Rapid (results used in a diagnosis of a patient or as a means of ensuring the safety of a process, usually require the test result to be provided as quickly as possible)
Robust (this is a catch-all term: simply put, the test protocol should be simple enough to be carried out reliably under the conditions where it is most likely to be applied. For example, if the test works reliably only in an hermetically sealed laboratory, with specialised equipment, it is unlikely to be suitable for use in "the field")
Economical (this is a relative term, but I believe students should be aware of the cost of experimental instruments and reagents. In the case of a complex clinical diagnosis, the cost per test has to be commensurate with the level of funds available to the health provider (eg the NHS).[This argument crosses over into the cost of drugs].
Simple (as one of world's greatest intellects, Leonardo da Vinci, once said: "Simplicity is the ultimate sophistication." In my own experience, you know when you haven't quite got a method or an idea correct: it lacks the simplicity of the most enduring concepts and methods)
 Let me take you through a method that we all rely on to ensure our water is safe to drink: and NHS advice is to drink just over 1 litre of water per day. [If everyone in the world did drink this amount, the total volume of water consumed per day would simply be the world population figure (7.8bn) in litres per day. Sadly, this number is presumably significantly less than this.] Randomly selecting one test, I have chosen to describe a colourimetric test for the estimation of (toxic) mercury ions in water supplies.
Let me take you through a method that we all rely on to ensure our water is safe to drink: and NHS advice is to drink just over 1 litre of water per day. [If everyone in the world did drink this amount, the total volume of water consumed per day would simply be the world population figure (7.8bn) in litres per day. Sadly, this number is presumably significantly less than this.] Randomly selecting one test, I have chosen to describe a colourimetric test for the estimation of (toxic) mercury ions in water supplies. A consortium of European Laboratories in Spain, Switzerland and the UK, recently developed a method for the detection of mercury ions, based on a colour change that makes the detection of submicromolar levels of mercury possible. It is not the only method, but it is one that provides a benchmark for all tests and you can read more here in the abstract, if you are interested. The principle of the method is that the coordination of mercury (Hg) ions by a dye known as ruthenium complex N719 [bis(2,2‘-bipyridyl-4,4‘-dicarboxylate)ruthenium(II) bis(tetrabutylammonium) bis(thiocyanate)](sorry!) changes the colour of the dye from green to purple. The structure of the dye is shown above on the left: the two sites labelled NCS (non-coordinated) are the points of attachment of the Hg ions, and the aromatic nature of the ruthenium complex provides the electrons that lead to selective absorption of visible light upon addition of the right metal ion. As you can see below, the addition of no metal ions or a range of typical contaminating metals gives a clear test of the presence of mercury ions at concentrations as low as 0.013ppm.
 |
| From left to right: no metal ions, Hg2+, Cd2+, Pb2+, Fe2+, Cu2+, and Zn2+ |
Sensitivity The acceptable level of mercury ions in water supplies is less than 0.02ppm. So this method meets the criteria, but just!
Specificity (or sometimes selectivity). The published data show that the method can easily distinguish between the closely related metal ions.
Rapid There is a spontaneous (within seconds) colour change when the reagent (the dye) is added to the sample: so all good in this respect. In fact the method can be adapted to make it similar to a litmus test, by impregnating paper with the dye.
Robust This type of method can be carried out anywhere, subject to the appropriate safety measures and access to reagents (the amounts of the dye may be limiting).
Economical The cost of reagents and materials is at the very low end and therefor this method is very inexpensive
Simple In the case of the dipstick type method, it couldn't be easier. The use of a simple visible light colourimeter to obtain accurate numbers is at the very low end of instrument costs and this method could easily be adapted for smart-phone detection.
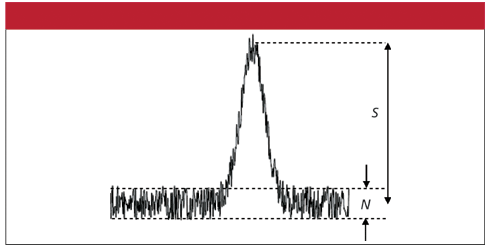
In short, this method meets all the criteria well, but the limit of detection may need to be improved in certain situations. I am referring here to the so called signal to noise ratio (SNR). The figure on the right illustrates the SNR in the case of a spectral measurement. This is an example of a good SNR, where the SNR is much greater than 1. A poor SNR occurs when this ratio is less than or equal to 1. The SNR can be improved by electronic or optical improvements, but remember this will always tend to reduce the simplicity and increase the cost of a test.
Let me turn now to the types of tests that are being used as part of the worldwide strategy for dealing with Covid 19
There are two technologies that need an explanation. The first is the Polymerase Chain Reaction (PCR) and the second is immuno-detection. The first is at the core of the detection of infection by the virus, while the second is used to detect the presence of the virus (in this case) or antibodies raised against the virus by an individual in recovery. These methodologies have emerged largely from HIV and influenza clinical and diagnostic research. I shall use the criteria above to explain the limitations of each of the methods and then finally, I shall provide some links to further publications and commercial sites.
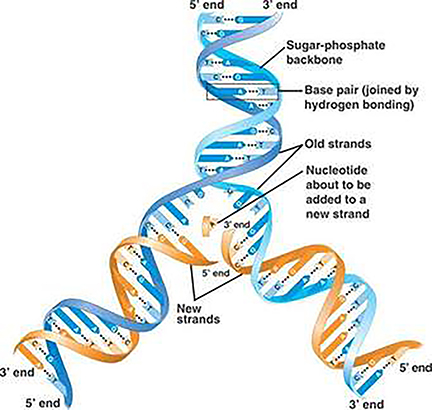
1. The PCR Test Viral detection by amplification based on PCR involves obtaining a sample of virus from a patient. Typically, in the case of Covid-19, the virus is recovered by swabbing the back of the throat and quickly returning the swab to a sterile tube for analysis. [I shall not comment here on best clinical practice and risks etc. I have no experience of clinical collection of this type]. The virus has a 30 000 bp genome comprising RNA. However, the PCR requires DNA as the substrate (or template) for the reaction to proceed. In the case of bacterial infections, PCR can be used directly on swabs, but an additional step is required for the detection of Covid-19 (and HIV/influenza). This step involves the accurate conversion of the RNA genome (or sections of it) into DNA (often called copy, or cDNA). Reverse Transcriptase (RT) enzymes are characteristic constituents of certain classes of RNA viruses, like HIV: these are called retroviruses, these enzymes are used to carry out the conversion step.
 One of the biggest challenges to a successful RT reaction is not the quality of the enzyme (although for research this may be the case), but rather the quality and quantity of the RNA template, and the exposure of the sample to enzymes called ribonucleases, which are notoriously robust and ubiquitous. These enzymes will degrade the extracted RNA, making downstream testing a major challenge. The methodology for extracting and stabilizing RNA has remained largely unchanged for over thirty years and is an important (if somewhat mundane) area for improvement. The components of the method described by Piotr Chomczynski and Nicolett Sacchi comprises a cocktail of guanidinium thiocyanate (a hazardous general denaturant), sodium citrate (a tricarboxylic acid salt) and sarcosyl (a negatively charged, mild detergent, often added to soap and shampoo) and a second solvent often incorporating phenol-chloroform. There are many manufacturers, but the availability of such a key set of reagents may be proving a challenge for some countries where there is a reliance on overseas supply chains. As a result of mixing biological samples from the swab (cells, virus and mucus), the released viral RNA can then be converted to DNA by a polymerization reaction comprising the enzyme (RT), the nucleotide precursors of DNA (dATP, dGTP, dCTP and dTTP: remember the RNA contains the ribose form of nucleotide, but DNA the deoxy (d) form). A suitable biological buffer and a primer that is a short synthetic sequence of DNA complementary to a specific region of the viral genome (this gives the test its specificity).The reaction is illustrated above, the primer is added at the first polymerization step. The double stranded DNA, represents a faithful copy of the RNA genome and is now ready for the second step, which produces the all important readout.
One of the biggest challenges to a successful RT reaction is not the quality of the enzyme (although for research this may be the case), but rather the quality and quantity of the RNA template, and the exposure of the sample to enzymes called ribonucleases, which are notoriously robust and ubiquitous. These enzymes will degrade the extracted RNA, making downstream testing a major challenge. The methodology for extracting and stabilizing RNA has remained largely unchanged for over thirty years and is an important (if somewhat mundane) area for improvement. The components of the method described by Piotr Chomczynski and Nicolett Sacchi comprises a cocktail of guanidinium thiocyanate (a hazardous general denaturant), sodium citrate (a tricarboxylic acid salt) and sarcosyl (a negatively charged, mild detergent, often added to soap and shampoo) and a second solvent often incorporating phenol-chloroform. There are many manufacturers, but the availability of such a key set of reagents may be proving a challenge for some countries where there is a reliance on overseas supply chains. As a result of mixing biological samples from the swab (cells, virus and mucus), the released viral RNA can then be converted to DNA by a polymerization reaction comprising the enzyme (RT), the nucleotide precursors of DNA (dATP, dGTP, dCTP and dTTP: remember the RNA contains the ribose form of nucleotide, but DNA the deoxy (d) form). A suitable biological buffer and a primer that is a short synthetic sequence of DNA complementary to a specific region of the viral genome (this gives the test its specificity).The reaction is illustrated above, the primer is added at the first polymerization step. The double stranded DNA, represents a faithful copy of the RNA genome and is now ready for the second step, which produces the all important readout.
The PCR reaction comprises 3 phases, all carried out in a typical volume of 0.01ml in a thin-walled plastic tube in an instrument simply referred to as a PCR machine, or by others as an intelligent heating block! The schematic diagram from New England Biolabs shows the key stages below.
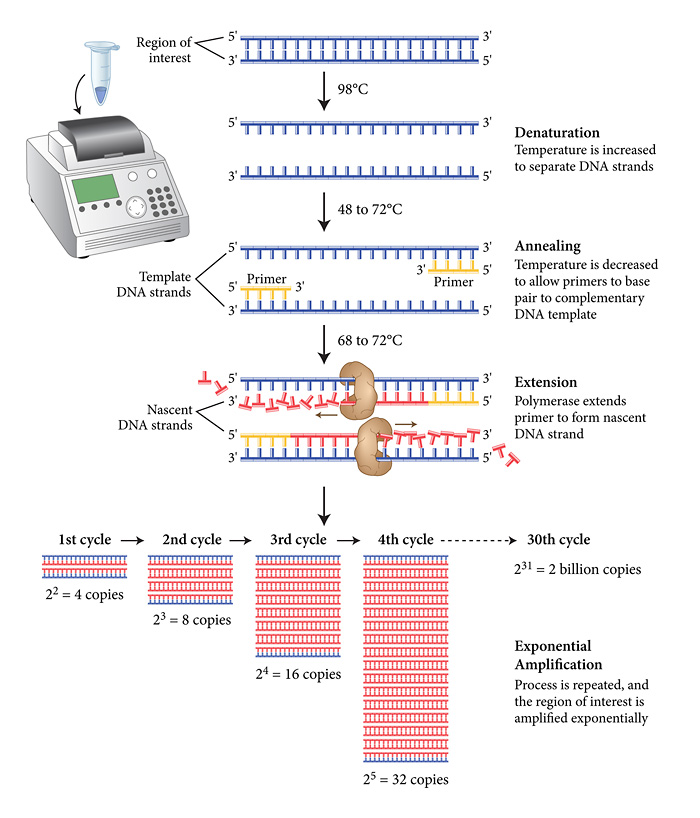 The viral cDNA is first denatured into two complementary single strands at around the boiling point of water. A set of primers is then added (in vast molar excess) along with the dNTP precursors (also in molar excess) together with a thermostable DNA polymerase (to my knowledge the last UK site to manufacture these enzymes (apart from academic research labs) moved offshore a couple of years ago). As you may have noticed, the extension reaction proceeds at relatively high temperatures (around 70 degrees Celsius) to ensure the template strands remain separate as the conversion of the template proceeds. In addition, the final step is followed by a second (third, fourth etc) denaturing step at almost 100 degrees Celsius. This places a stress on the active "life" of the enzymes, but thankfully, Biodiversity came to the rescue 40 years ago with the discovery of thermostable and now hyper-thermostable DNA polymerases (you may have heard of Taq, Pfu polymerases, among others). As illustrated above there is an exponential increase in the amount of DNA produced during the PCR. The progress of this amplification reaction is exactly the same mathematically as discussed in my previous post. In a typical reaction, 25-30 cycles are carried out and the length of time taken for the PCR is between 20 minutes (the best reagents and instruments) and 4 hours.
The viral cDNA is first denatured into two complementary single strands at around the boiling point of water. A set of primers is then added (in vast molar excess) along with the dNTP precursors (also in molar excess) together with a thermostable DNA polymerase (to my knowledge the last UK site to manufacture these enzymes (apart from academic research labs) moved offshore a couple of years ago). As you may have noticed, the extension reaction proceeds at relatively high temperatures (around 70 degrees Celsius) to ensure the template strands remain separate as the conversion of the template proceeds. In addition, the final step is followed by a second (third, fourth etc) denaturing step at almost 100 degrees Celsius. This places a stress on the active "life" of the enzymes, but thankfully, Biodiversity came to the rescue 40 years ago with the discovery of thermostable and now hyper-thermostable DNA polymerases (you may have heard of Taq, Pfu polymerases, among others). As illustrated above there is an exponential increase in the amount of DNA produced during the PCR. The progress of this amplification reaction is exactly the same mathematically as discussed in my previous post. In a typical reaction, 25-30 cycles are carried out and the length of time taken for the PCR is between 20 minutes (the best reagents and instruments) and 4 hours.
I need to now add an extra step that allows the operator to extract even
 more information and to observe the outcome PCR in real time. By adding a fluorescent reagent into the reaction mixture, in a way that leads to an increase in signal as the amount of amplified DNA (sometimes referred to as the amplicon) increases, it is possible to obtain a real time progress trace of the reaction. In the past, and still for many applications, scientists detect the "end-point" of the reaction, but "real-time" or "quantitative" PCR is the Gold-Standard for diagnostic scrutiny in a clinical setting. There are several ways in which fluorescence can be harnessed. An additional single strand of DNA complementary to the template can be added. This "probe" will anneal to its complementary sequence (but is chemically designed not to act as a primer) and in its duplex form it may have no, or a very low fluorescence. However, as it is displaced during the extension phase, the previously quenched fluorophores now emit a signal which can be detected, as shown in the trace above). In another approach, small molecule fluorophores that differentially emit in the free (in solution) and bound (complexed with double-stranded DNA) states, can be used for QPCR. For those who want a more detailed explanation of QPCR methods take a look at the Wiki page here, to get you started and here for the more inquisitive.
more information and to observe the outcome PCR in real time. By adding a fluorescent reagent into the reaction mixture, in a way that leads to an increase in signal as the amount of amplified DNA (sometimes referred to as the amplicon) increases, it is possible to obtain a real time progress trace of the reaction. In the past, and still for many applications, scientists detect the "end-point" of the reaction, but "real-time" or "quantitative" PCR is the Gold-Standard for diagnostic scrutiny in a clinical setting. There are several ways in which fluorescence can be harnessed. An additional single strand of DNA complementary to the template can be added. This "probe" will anneal to its complementary sequence (but is chemically designed not to act as a primer) and in its duplex form it may have no, or a very low fluorescence. However, as it is displaced during the extension phase, the previously quenched fluorophores now emit a signal which can be detected, as shown in the trace above). In another approach, small molecule fluorophores that differentially emit in the free (in solution) and bound (complexed with double-stranded DNA) states, can be used for QPCR. For those who want a more detailed explanation of QPCR methods take a look at the Wiki page here, to get you started and here for the more inquisitive.
I hope the methodology is reasonably clear now, but let's take a look at the methodology for RT-QPCR detection of Covid-19 in the context of our criteria above. In respect of sensitivity, the introduction of fluorescence technology ensures that the method it is possible to detect around 100 virus particles per ml. As ever, while this is about as good as it gets, 100 becomes 200 pretty quickly...With respect to specificity, anyone who uses PCR on a regular basis, knows that Watson-Crick base-pair rules, while theoretically sound, practically lead to false positives until the reaction conditions are fine-tuned. While this is a nuisance in research, commercial diagnostic kits on the market, will have ironed out these issues, and there is good scrutiny and recommendations available online from the testing community. So this is usually an early issue, that goes away pretty quickly. The early release of the Covid-19 RNA genome sequence by the Chinese scientific community, has made the design of primers, key to selective amplification of the genome possible. Is it rapid enough? The fastest turn-around times are typically a few hours from sampling to amplification, but sampling and delivery are often at separate sites. In addition, central testing labs are often asked to confirm that suspected positives are real, especially during the early stages of an outbreak. This can mean that the actual time for issuing a result can be up to a couple of days. This needs fixing! The robustness of PCR methods, which have been around in diagnostic labs for around 30 years (QPCR, 20) is seen to be satisfactory: the supply of reagents will mirror the supply chains of the automobile industry (precursors and primers are typically manufactured overseas and shipped on demand, although I assume bulk buying will be normal in diagnostic labs).
The economics of PCR methodology is a moot point. I don't want to get into the historical IP battles between Roche and the Biotech Community, but suffice to say, this is a big source of revenue for the commercial diagnostics sector and while prices are competitive, the manufacturing of reagents and instruments can yield high margins for some companies. I suspect the Covid-19 crisis might lead to a re-think of the sector? Finally, is it simple? PCR has become a run-of -the-mill technique in Biotechnology, but it is a multi-step process that has elements of simplicity and complexity. In essence we are harnessing a fundamental process of Nature, the replication of a genome. Personally, I would describe it as elegant, but sophisticated and in a separate post, just to whet your appetites, James (my PhD student) and I will reflect on the sophistication of the technology and the Molecular Biology of nucleic acid polymerases polymerases.
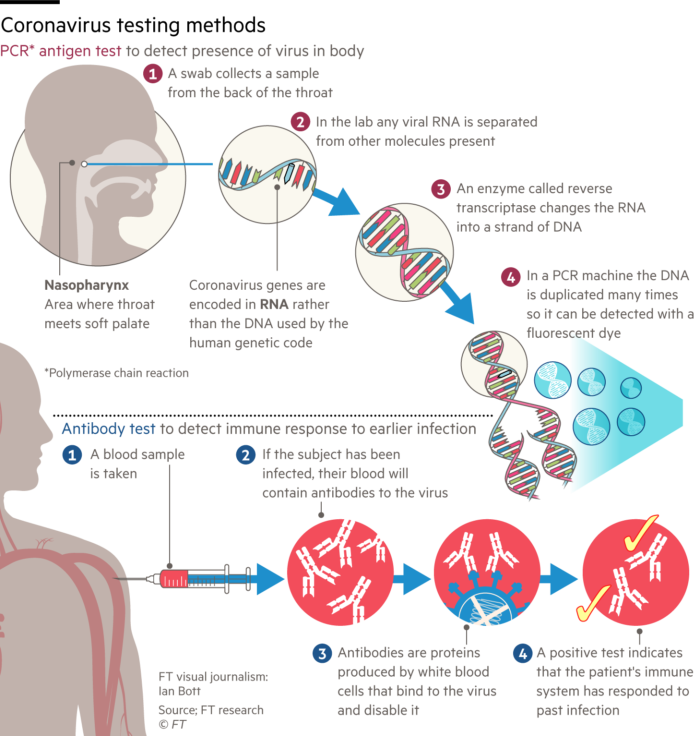 Methods that rely on the availability of antibodies for the detection of either viruses or the antibodies raised in response to an infection are the subject of this section. First I have to try and undo some of the confusion in the media. The Financial Times today published an open access article on the testing methodology. Reluctantly, I have included the online diagram on the left (I have deliberately reduced the size since it is misleading). The report describes two tests for Covid-19. The "PCR-antigen test" and the "antibody test". The combination of PCR with antigen is just so inappropriate. The diagram does illustrate the process of converting viral, genomic RNA into a fluorescent readout of DNA amplification by PCR, but the word antigen is misleading in this context. Let's now clarify: there are 3 possible virus tests:
Methods that rely on the availability of antibodies for the detection of either viruses or the antibodies raised in response to an infection are the subject of this section. First I have to try and undo some of the confusion in the media. The Financial Times today published an open access article on the testing methodology. Reluctantly, I have included the online diagram on the left (I have deliberately reduced the size since it is misleading). The report describes two tests for Covid-19. The "PCR-antigen test" and the "antibody test". The combination of PCR with antigen is just so inappropriate. The diagram does illustrate the process of converting viral, genomic RNA into a fluorescent readout of DNA amplification by PCR, but the word antigen is misleading in this context. Let's now clarify: there are 3 possible virus tests:
1. The PCR Test (as discussed above)
2. The Antibody Test (below)
3. The Antigen Test (also below).
2. The Antibody Test is based on the identification of antibodies (immunoglobulins) developed by an individual in response to a Covid-19 infection. This test is used mainly to establish whether an individual has recovered from an infection. The test can be carried out any time post recovery, but is particularly useful to establish whether someone is likely to have developed immunity to a second infection from the same virus. Under the current circumstances in the case of say a member of the NHS, this information helps to recommend a return to work.
 Following any viral infection, a healthy individual develops a repertoire of antibodies to the infective agent, in this case the surface of the Covid- 19 virus, the spike protein. Physiological antibody responses are typically polyclonal, whereas antibodies used in diagnostics and therapeutics are generally monoclonal. These terms are used widely in medicine and medical science, but for these purposes the definition is quite simple. Imagine a spike protein as shown on the left. Four antibodies (not to scale) are shown binding to different locations on the surface of the spike antigen. A monoclonal antibody event would be illustrated by (for example) just the yellow antibody, whereas a polyclonal antibody response leads to a spectrum of yellow, purple, red and green antibodies covering different surface patches (or epitopes). Clearly if you are trying to eliminate an antigen, such as a virus, it is better to generate multiple antibody types. If on the other hand you are looking for selectivity and recognition of a unique epitope, a monoclonal antibody may be better, especially for therapeutics and for in vitro diagnostics.
Following any viral infection, a healthy individual develops a repertoire of antibodies to the infective agent, in this case the surface of the Covid- 19 virus, the spike protein. Physiological antibody responses are typically polyclonal, whereas antibodies used in diagnostics and therapeutics are generally monoclonal. These terms are used widely in medicine and medical science, but for these purposes the definition is quite simple. Imagine a spike protein as shown on the left. Four antibodies (not to scale) are shown binding to different locations on the surface of the spike antigen. A monoclonal antibody event would be illustrated by (for example) just the yellow antibody, whereas a polyclonal antibody response leads to a spectrum of yellow, purple, red and green antibodies covering different surface patches (or epitopes). Clearly if you are trying to eliminate an antigen, such as a virus, it is better to generate multiple antibody types. If on the other hand you are looking for selectivity and recognition of a unique epitope, a monoclonal antibody may be better, especially for therapeutics and for in vitro diagnostics.
The question is, how do you recognise one or more of the antibodies that an
 individual is likely to have in his/her blood stream following an infection? There are several ways, but essentially, if you present a recombinant form of say the spike protein (blue circle on the right), or an inactivated preparation of the virus itself, to a sample of blood, any virus-specific antibodies, if present, will bind to the virus (or parts thereof) and using a second antibody, that detects all human immunoglobulins, carrying a fluorescent dye or a detectable enzyme, the presence of the antibodies can be detected. This technology has been widely employed in the management and epidemiology of HIV infection and is shown schematically on the right, where the blue secondary antibody has an enzyme attached (green ball) which catalyses the conversion of a substrate into a detectable product (blue sun). I shall combine an evaluation of this test and the Antigen Test below.
individual is likely to have in his/her blood stream following an infection? There are several ways, but essentially, if you present a recombinant form of say the spike protein (blue circle on the right), or an inactivated preparation of the virus itself, to a sample of blood, any virus-specific antibodies, if present, will bind to the virus (or parts thereof) and using a second antibody, that detects all human immunoglobulins, carrying a fluorescent dye or a detectable enzyme, the presence of the antibodies can be detected. This technology has been widely employed in the management and epidemiology of HIV infection and is shown schematically on the right, where the blue secondary antibody has an enzyme attached (green ball) which catalyses the conversion of a substrate into a detectable product (blue sun). I shall combine an evaluation of this test and the Antigen Test below.
 3. The Antigen Test is used to detect the presence of a Covid-19 protein, such as the spike protein. This test follows very similar principles to the Antibody Test, in that it relies on the strong and selective binding between an antibody and an antigen and the ability to couple a fluorescent or enzymatic detection unit to, in this case the antibody component. The first requirement is the production of a monoclonal antibody (usually) to a preparation of the virus, or more typically the spike protein. There are several ways to generate a monoclonal antibody, but either way it takes around 2-3 months to do so. This antibody would then be coupled to the detection unit and used to interrogate patient samples, in this case it could be nasal extracts or blood samples. If there is sufficient virus in circulation, the spike-specific antibody will bind to it, and as above the signal can be detected by comparing the level of colour or fluorescence with negative samples. Recently a number of companies have launched Covid-19 tests of this type: I have taken an image (top left) form the Corisbio web site to illustrate their kit which is remarkably similar to a pregnancy test kit. In fact the pregnancy kits which have been available for over 40 years are based on the recognition of elevated levels of chorionic gonadotropin in the urine, in a very similar way.
3. The Antigen Test is used to detect the presence of a Covid-19 protein, such as the spike protein. This test follows very similar principles to the Antibody Test, in that it relies on the strong and selective binding between an antibody and an antigen and the ability to couple a fluorescent or enzymatic detection unit to, in this case the antibody component. The first requirement is the production of a monoclonal antibody (usually) to a preparation of the virus, or more typically the spike protein. There are several ways to generate a monoclonal antibody, but either way it takes around 2-3 months to do so. This antibody would then be coupled to the detection unit and used to interrogate patient samples, in this case it could be nasal extracts or blood samples. If there is sufficient virus in circulation, the spike-specific antibody will bind to it, and as above the signal can be detected by comparing the level of colour or fluorescence with negative samples. Recently a number of companies have launched Covid-19 tests of this type: I have taken an image (top left) form the Corisbio web site to illustrate their kit which is remarkably similar to a pregnancy test kit. In fact the pregnancy kits which have been available for over 40 years are based on the recognition of elevated levels of chorionic gonadotropin in the urine, in a very similar way.
The antibody and antigen test kits all rely on the tried
 and tested technology associated with antibody: antigen recognition and the associated reporter technologies. There is no doubt that they are sufficiently, sensitive, specific, robust and are faster to perform than PCR methods. However, they are completely dependent on the high affinity and selectivity of the antibody/antigen preparations (an the human physiological responses). If you look at HIV testing, as I mentioned with vaccines, this remains a work in progress, and I expect this will be the case with both Antibody and Antigen tests: they will perform an important function, but they will be sub-optimal at first, improving over the coming years
and tested technology associated with antibody: antigen recognition and the associated reporter technologies. There is no doubt that they are sufficiently, sensitive, specific, robust and are faster to perform than PCR methods. However, they are completely dependent on the high affinity and selectivity of the antibody/antigen preparations (an the human physiological responses). If you look at HIV testing, as I mentioned with vaccines, this remains a work in progress, and I expect this will be the case with both Antibody and Antigen tests: they will perform an important function, but they will be sub-optimal at first, improving over the coming years
UPDATE
 After my earlier post, in which I looked at the underlying principles behind diagnostic testing, the controversy lingers on. This week, the slightly embattled Health Secretary, Mr. Matt Hancock, announced the UK would be carrying out 100 000 individual tests per day by the end of the month. However, this was reported in a longer article The Guardian as follows:
After my earlier post, in which I looked at the underlying principles behind diagnostic testing, the controversy lingers on. This week, the slightly embattled Health Secretary, Mr. Matt Hancock, announced the UK would be carrying out 100 000 individual tests per day by the end of the month. However, this was reported in a longer article The Guardian as follows:
Matt Hancock, the health secretary, talked with confidence of achieving 25,000 virus tests a day for those ill in hospital and NHS staff who have symptoms or are in a household with somebody who is sick, so that they can go back to work. These are PCR (polymerase chain reaction) swab tests, sometimes called antigen tests, which need to be processed with specialised kits in a lab.
Yes, the mixing of immunology and molecular genetics once again! But I wanted to just make sure that you understand the current challenges that face those trying to deliver the necessary materials for the tests, which comprise:
The second point I want to raise is the value of the tests, by which I mean, how will testing on this scale help us beat the virus? The test to establish whether an individual is infected by Covid-19 or any diagnostic test, determines how you manage that person's recovery and also how best you protect those who have been in close contact. In an ideal world, where a "cure" for Covid-19 was available, an individual would be given the medicine, either to take home or as part of a short stay in hospital. This would be the case for many heart diseases, for example. In the absence of a cure for Covid-19, the treatment is largely a matter of support based on the patient's age, general mental and physical state and underlying health conditions. Essentially, the patient is nursed as s/he marshals her/his immune system to fight off the viral infection.
There are two technologies that need an explanation. The first is the Polymerase Chain Reaction (PCR) and the second is immuno-detection. The first is at the core of the detection of infection by the virus, while the second is used to detect the presence of the virus (in this case) or antibodies raised against the virus by an individual in recovery. These methodologies have emerged largely from HIV and influenza clinical and diagnostic research. I shall use the criteria above to explain the limitations of each of the methods and then finally, I shall provide some links to further publications and commercial sites.
1. The PCR Test Viral detection by amplification based on PCR involves obtaining a sample of virus from a patient. Typically, in the case of Covid-19, the virus is recovered by swabbing the back of the throat and quickly returning the swab to a sterile tube for analysis. [I shall not comment here on best clinical practice and risks etc. I have no experience of clinical collection of this type]. The virus has a 30 000 bp genome comprising RNA. However, the PCR requires DNA as the substrate (or template) for the reaction to proceed. In the case of bacterial infections, PCR can be used directly on swabs, but an additional step is required for the detection of Covid-19 (and HIV/influenza). This step involves the accurate conversion of the RNA genome (or sections of it) into DNA (often called copy, or cDNA). Reverse Transcriptase (RT) enzymes are characteristic constituents of certain classes of RNA viruses, like HIV: these are called retroviruses, these enzymes are used to carry out the conversion step.
One of the biggest challenges to a successful RT reaction is not the quality of the enzyme (although for research this may be the case), but rather the quality and quantity of the RNA template, and the exposure of the sample to enzymes called ribonucleases, which are notoriously robust and ubiquitous. These enzymes will degrade the extracted RNA, making downstream testing a major challenge. The methodology for extracting and stabilizing RNA has remained largely unchanged for over thirty years and is an important (if somewhat mundane) area for improvement. The components of the method described by Piotr Chomczynski and Nicolett Sacchi comprises a cocktail of guanidinium thiocyanate (a hazardous general denaturant), sodium citrate (a tricarboxylic acid salt) and sarcosyl (a negatively charged, mild detergent, often added to soap and shampoo) and a second solvent often incorporating phenol-chloroform. There are many manufacturers, but the availability of such a key set of reagents may be proving a challenge for some countries where there is a reliance on overseas supply chains. As a result of mixing biological samples from the swab (cells, virus and mucus), the released viral RNA can then be converted to DNA by a polymerization reaction comprising the enzyme (RT), the nucleotide precursors of DNA (dATP, dGTP, dCTP and dTTP: remember the RNA contains the ribose form of nucleotide, but DNA the deoxy (d) form). A suitable biological buffer and a primer that is a short synthetic sequence of DNA complementary to a specific region of the viral genome (this gives the test its specificity).The reaction is illustrated above, the primer is added at the first polymerization step. The double stranded DNA, represents a faithful copy of the RNA genome and is now ready for the second step, which produces the all important readout. The PCR reaction comprises 3 phases, all carried out in a typical volume of 0.01ml in a thin-walled plastic tube in an instrument simply referred to as a PCR machine, or by others as an intelligent heating block! The schematic diagram from New England Biolabs shows the key stages below.
I need to now add an extra step that allows the operator to extract even

I hope the methodology is reasonably clear now, but let's take a look at the methodology for RT-QPCR detection of Covid-19 in the context of our criteria above. In respect of sensitivity, the introduction of fluorescence technology ensures that the method it is possible to detect around 100 virus particles per ml. As ever, while this is about as good as it gets, 100 becomes 200 pretty quickly...With respect to specificity, anyone who uses PCR on a regular basis, knows that Watson-Crick base-pair rules, while theoretically sound, practically lead to false positives until the reaction conditions are fine-tuned. While this is a nuisance in research, commercial diagnostic kits on the market, will have ironed out these issues, and there is good scrutiny and recommendations available online from the testing community. So this is usually an early issue, that goes away pretty quickly. The early release of the Covid-19 RNA genome sequence by the Chinese scientific community, has made the design of primers, key to selective amplification of the genome possible. Is it rapid enough? The fastest turn-around times are typically a few hours from sampling to amplification, but sampling and delivery are often at separate sites. In addition, central testing labs are often asked to confirm that suspected positives are real, especially during the early stages of an outbreak. This can mean that the actual time for issuing a result can be up to a couple of days. This needs fixing! The robustness of PCR methods, which have been around in diagnostic labs for around 30 years (QPCR, 20) is seen to be satisfactory: the supply of reagents will mirror the supply chains of the automobile industry (precursors and primers are typically manufactured overseas and shipped on demand, although I assume bulk buying will be normal in diagnostic labs).
The economics of PCR methodology is a moot point. I don't want to get into the historical IP battles between Roche and the Biotech Community, but suffice to say, this is a big source of revenue for the commercial diagnostics sector and while prices are competitive, the manufacturing of reagents and instruments can yield high margins for some companies. I suspect the Covid-19 crisis might lead to a re-think of the sector? Finally, is it simple? PCR has become a run-of -the-mill technique in Biotechnology, but it is a multi-step process that has elements of simplicity and complexity. In essence we are harnessing a fundamental process of Nature, the replication of a genome. Personally, I would describe it as elegant, but sophisticated and in a separate post, just to whet your appetites, James (my PhD student) and I will reflect on the sophistication of the technology and the Molecular Biology of nucleic acid polymerases polymerases.
Methods that rely on the availability of antibodies for the detection of either viruses or the antibodies raised in response to an infection are the subject of this section. First I have to try and undo some of the confusion in the media. The Financial Times today published an open access article on the testing methodology. Reluctantly, I have included the online diagram on the left (I have deliberately reduced the size since it is misleading). The report describes two tests for Covid-19. The "PCR-antigen test" and the "antibody test". The combination of PCR with antigen is just so inappropriate. The diagram does illustrate the process of converting viral, genomic RNA into a fluorescent readout of DNA amplification by PCR, but the word antigen is misleading in this context. Let's now clarify: there are 3 possible virus tests:1. The PCR Test (as discussed above)
2. The Antibody Test (below)
3. The Antigen Test (also below).
2. The Antibody Test is based on the identification of antibodies (immunoglobulins) developed by an individual in response to a Covid-19 infection. This test is used mainly to establish whether an individual has recovered from an infection. The test can be carried out any time post recovery, but is particularly useful to establish whether someone is likely to have developed immunity to a second infection from the same virus. Under the current circumstances in the case of say a member of the NHS, this information helps to recommend a return to work.
 Following any viral infection, a healthy individual develops a repertoire of antibodies to the infective agent, in this case the surface of the Covid- 19 virus, the spike protein. Physiological antibody responses are typically polyclonal, whereas antibodies used in diagnostics and therapeutics are generally monoclonal. These terms are used widely in medicine and medical science, but for these purposes the definition is quite simple. Imagine a spike protein as shown on the left. Four antibodies (not to scale) are shown binding to different locations on the surface of the spike antigen. A monoclonal antibody event would be illustrated by (for example) just the yellow antibody, whereas a polyclonal antibody response leads to a spectrum of yellow, purple, red and green antibodies covering different surface patches (or epitopes). Clearly if you are trying to eliminate an antigen, such as a virus, it is better to generate multiple antibody types. If on the other hand you are looking for selectivity and recognition of a unique epitope, a monoclonal antibody may be better, especially for therapeutics and for in vitro diagnostics.
Following any viral infection, a healthy individual develops a repertoire of antibodies to the infective agent, in this case the surface of the Covid- 19 virus, the spike protein. Physiological antibody responses are typically polyclonal, whereas antibodies used in diagnostics and therapeutics are generally monoclonal. These terms are used widely in medicine and medical science, but for these purposes the definition is quite simple. Imagine a spike protein as shown on the left. Four antibodies (not to scale) are shown binding to different locations on the surface of the spike antigen. A monoclonal antibody event would be illustrated by (for example) just the yellow antibody, whereas a polyclonal antibody response leads to a spectrum of yellow, purple, red and green antibodies covering different surface patches (or epitopes). Clearly if you are trying to eliminate an antigen, such as a virus, it is better to generate multiple antibody types. If on the other hand you are looking for selectivity and recognition of a unique epitope, a monoclonal antibody may be better, especially for therapeutics and for in vitro diagnostics. The question is, how do you recognise one or more of the antibodies that an
 3. The Antigen Test is used to detect the presence of a Covid-19 protein, such as the spike protein. This test follows very similar principles to the Antibody Test, in that it relies on the strong and selective binding between an antibody and an antigen and the ability to couple a fluorescent or enzymatic detection unit to, in this case the antibody component. The first requirement is the production of a monoclonal antibody (usually) to a preparation of the virus, or more typically the spike protein. There are several ways to generate a monoclonal antibody, but either way it takes around 2-3 months to do so. This antibody would then be coupled to the detection unit and used to interrogate patient samples, in this case it could be nasal extracts or blood samples. If there is sufficient virus in circulation, the spike-specific antibody will bind to it, and as above the signal can be detected by comparing the level of colour or fluorescence with negative samples. Recently a number of companies have launched Covid-19 tests of this type: I have taken an image (top left) form the Corisbio web site to illustrate their kit which is remarkably similar to a pregnancy test kit. In fact the pregnancy kits which have been available for over 40 years are based on the recognition of elevated levels of chorionic gonadotropin in the urine, in a very similar way.
3. The Antigen Test is used to detect the presence of a Covid-19 protein, such as the spike protein. This test follows very similar principles to the Antibody Test, in that it relies on the strong and selective binding between an antibody and an antigen and the ability to couple a fluorescent or enzymatic detection unit to, in this case the antibody component. The first requirement is the production of a monoclonal antibody (usually) to a preparation of the virus, or more typically the spike protein. There are several ways to generate a monoclonal antibody, but either way it takes around 2-3 months to do so. This antibody would then be coupled to the detection unit and used to interrogate patient samples, in this case it could be nasal extracts or blood samples. If there is sufficient virus in circulation, the spike-specific antibody will bind to it, and as above the signal can be detected by comparing the level of colour or fluorescence with negative samples. Recently a number of companies have launched Covid-19 tests of this type: I have taken an image (top left) form the Corisbio web site to illustrate their kit which is remarkably similar to a pregnancy test kit. In fact the pregnancy kits which have been available for over 40 years are based on the recognition of elevated levels of chorionic gonadotropin in the urine, in a very similar way.The antibody and antigen test kits all rely on the tried
UPDATE
After my earlier post, in which I looked at the underlying principles behind diagnostic testing, the controversy lingers on. This week, the slightly embattled Health Secretary, Mr. Matt Hancock, announced the UK would be carrying out 100 000 individual tests per day by the end of the month. However, this was reported in a longer article The Guardian as follows:Matt Hancock, the health secretary, talked with confidence of achieving 25,000 virus tests a day for those ill in hospital and NHS staff who have symptoms or are in a household with somebody who is sick, so that they can go back to work. These are PCR (polymerase chain reaction) swab tests, sometimes called antigen tests, which need to be processed with specialised kits in a lab.
Yes, the mixing of immunology and molecular genetics once again! But I wanted to just make sure that you understand the current challenges that face those trying to deliver the necessary materials for the tests, which comprise:
Enzymes (polymerases to amplify the viral RNA and turn it into DNA)
Solvents (phenol, chloroform etc to separate proteins, lipids and nucleic acids)
Salts (buffers and stabilisers)
Fine chemicals (nucleotides and the reagents for synthesising DNA (phosphoramidites and the appropriate solvents etc).
The majority of these materials are typically imported from the EU, North America and East Asia. There is no doubt that the small labs (the University and Research Institute Labs that Sir Paul Nurse referred to as the equivalent of the Dunkirk little ships flotilla) have the capability and capacity to produce enzymes and to synthesise some of the "primers" needed for PCR, but the core reagents and solvents will require re-purposing some of our larger Chemical Industry Sites and possibly shifting synthesis priorities at some specialist Chemistry/Pharmaceutical manufacturers. I think we may reflect on our Global outsource models for maintaining the integrity of some of our supply chains in the aftermath of Covid-19?
 |
| Diagnosing Heart Disease with an ECG |
This is an important issue in medicine, that all of you considering any form of career in the healthcare sector should think about: the fact is that diagnosis often comes before a cure. Take the case of the breast cancer associated genes, BRCA1 and BRCA2 where certain mutations can increase the risk of contracting breast cancer. Pretty soon after the genes were discovered, diagnostic tests became available and many women were tested. In the months and years after, the tests became increasingly common. However, the knowledge that an individual carrying specific mutations in these genes was at a higher risk of developing early onset breast cancer became very sensitive. In the absence of a cure, the knowledge that you are at higher than normal risk can be liberating for some and devastating for others, from personal family experience. We need to understand that the value of testing for a disease that cannot (yet) be cured, must be fully explained. In the case of viral testing is an aid to developing management strategies for minimising the scale and rate of spread of the outbreak, it is also a means of increasing the accuracy of predictions and forecasts.
There has a considerable amount of debate around who should be tested. Some argue the healthcare professionals ranging from those on intensive care wards, where patients are experiencing life threatening symptoms, to those who argue patients should come first. Basically, we need to have the capability of working through the population as quickly as we can with the priority individuals identified as being critical for reducing the impact of the pandemic.
There has a considerable amount of debate around who should be tested. Some argue the healthcare professionals ranging from those on intensive care wards, where patients are experiencing life threatening symptoms, to those who argue patients should come first. Basically, we need to have the capability of working through the population as quickly as we can with the priority individuals identified as being critical for reducing the impact of the pandemic.